Solution: BIOVIA Discovery studio – Macromolecule tools
Benefits:
- -Reatailvely east technique
- -Less time to learn, do calculations and obtain results
- -Single and multiple template modelling
- -Pharmaceutically significant for drug screening.
CHALLENGE: Modelling of Nuclear protein of pharmaceutical interest
This customer utilized the Homology Modelling tool of BIOVIA Discovery Studio for building the homology model of the nuclear protein. The Modelling of nuclear receptor protein needs precision skills starting from its alignment with multiple templates in databases to its validation. Homology modelling is one of the key discoveries that led to a rapid paradigm shift in the field of computational biology.
Homology modelling obtains three-dimensional structure of a target protein based on the similarity between template and target sequences and this technique proves to be efficient in understanding membrane proteins that are hard to crystallize as it provides a higher degree of understanding of receptor-ligand interaction.
Homology modelling is a crucial method of computational biology, that uses structural biology concepts to determine the 3D structure of proteins. Since there was limited structural information or many nuclear protein structures were not available in the structural database repository led to the daunting task of resolving computationally. However, this customer has sound knowledge of pharmaceutical research and nuclear modelling and utilized the BIOVIA Discovery Studio macromolecule tools to address this issue with less effort.
Conditions followed for modelling
- 1) The length of the sequence must be 200-850 amino acids (This is mainly to obtain the best templates.)
- 2) The template identity must be >60% for single template modelling and >45% for multiple template modelling.
- 3) The alignment must have fewer gaps or no gaps
- 4) There should be least positives and hit crystallization <2. 5-3Å
RESULT: Computational homology modeling is equally comparable with experimental structures
The query protein sequence must hit at least 40%-60% identified with template structures.
Template identification is the critical first step

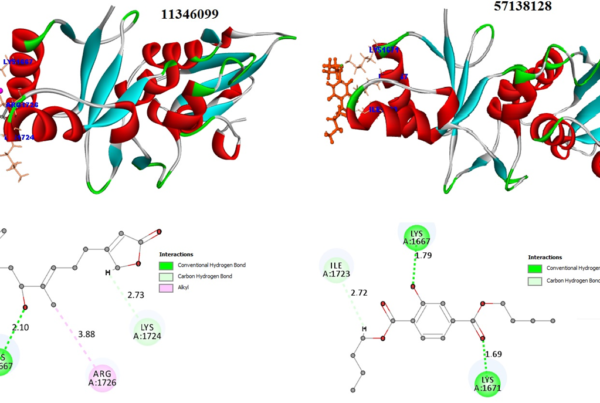
Using BLAST and PSI-BLAST programs, the target template(2ZK0) was identified. The target sequence is then aligned with the template structure(s) using the align sequence to template tool, which displays maximum sequence identity and similarity.

Further, the overhangs at N and C terminals are removed to improve the quality of the final model. Finally, using MODELLER, the final model was built using the built-in homology modelling tool.
The MODELLER has been integrated into the Discovery Studio for more than two decades. This tool was developed by Dr. Andrej Sal, UCSF, USA (https://salilab.org/modeller/).
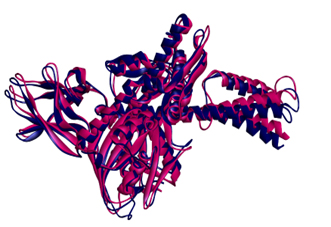
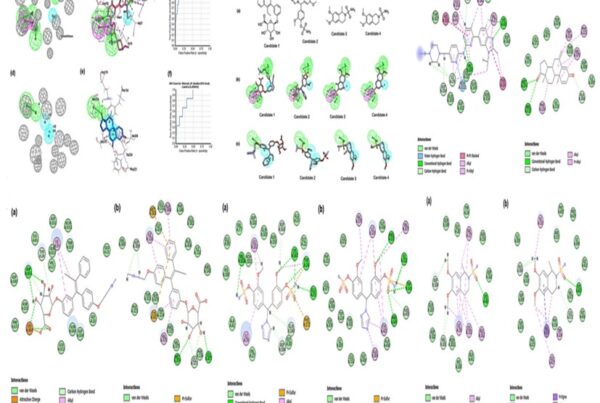
The final model obtained from the modeler shows the statistical quality and discrete optimised protein energy (DOPE) of -59147.3 with a root mean square deviation <1.8 Å. These results show an intact structural relationship between the template and query sequences. Furthermore, the stereochemical quality and consistency of the generating model were assessed using Ramachandra Plot and Verify profiles-3D. The results for the modelled nuclear protein PPAR-Gamma are extremely favorable.
SOLUTION: DISCOVERY STUDIO protein modelling is an alternative solution for the crystallization of nuclear protein
Nuclear proteins are major biochemically classified drug targets in the pharmaceutical industry. However, due to a lack of experimental structure, the research on nuclear proteins steeps down. But using BIOVIA Discovery Studio’s homology modelling and its tools will trespass the researcher’s issues. Furthermore, the amino acids in the Ramachandran plot of the modelled structure are in allowed and generously allowed regions, demonstrating the best qualities of the model and the technique used for modelling.

Hence, homology modelling is a good alternative approach to design the new protein and a boon to the research and pharmaceutical domain
Acknowledgement
We, the team, thank Dr. Prabitha and Dr. B. R. Prashantha Kumar, Department of Pharmaceutical Chemistry at JSS College of Pharmacy, Mysuru, for providing their research case study.
To cite: Prabitha and B.R. Prashantha Kumar, Homology modelling of nuclear proteins—An alternative approach to X-ray Crystallography, 2022.
Publication Date : October, 2022