Challenge:
To identify the natural source-based inhibitors from endolichenic fungi to target the role of the kinase in cancer
Solution:
BIOVIA Discovery studio –Simulation and Pharamcophore tools
Benefits:
- Exploration of the biological system in a short span of time.
- Cost-effective technique before cell line study.
- Easy to replicate the in vivo environment using thermodynamic parameters.
- Hassle-free workflow and results interpretation.
CHALLENGE: To explore the Anticancer secondary metabolites from different endolichenic fungi as kinase inhibitors
This customer utilized molecular simulation and pharmacophore modules of BIOVIA Discovery Studio to understand stability and the real-time atomic movement of biological systems and functional moiety responsible for molecular interaction.
The recent advances in the in-silico approaches like ADMET predictor, pharmacophore, molecular docking, and molecular dynamic simulation have become promising techniques to screen and analyze large data sets of molecules against various drug targets in a cost-effective manner without interference from experimental methods.
This customer studied the docked complex from an external system as input to study the real-time environment of the receptor-ligand interaction as a function of time using molecular dynamic simulation. Discovery studio has user-friendly space for importing and exporting the data is not hindered the prospect to drive the process. Also, their perspective is to understand the energy changes over a time period. Further, the painstaking challenge to draw the conclusion using an integrated approach of dynamic outcome with molecular mechanics based on Poisson–Boltzmann surface area (MM-PBSA). Final insights into the identification of functional moiety inference in binding interaction.
RESULT: Significance of the computational study to understand the real-time environment of biological system
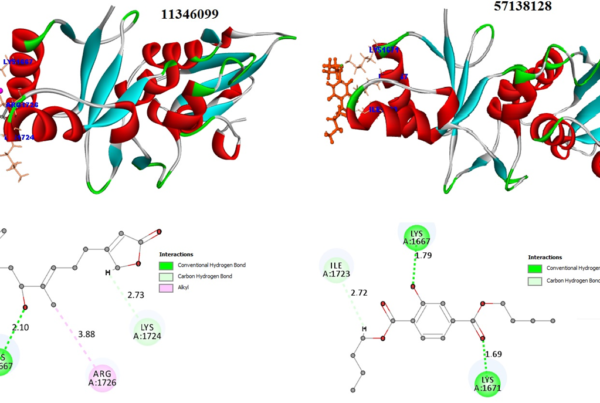
Natural-based compounds are usually toxic and free less side effects. However, the binding interaction and stability must be maintained for the best compounds to possess good biological activity. Molecular Dynamics simulation was performed with docked complexes involving 3D crystal structures of clinically important glycolytic kinases (human pyruvate kinase isoform 2 (3GQY), human hexokinase II (2NZT) and human phospoglycerokinase (4AXX)) bound with a natural metabolite of endolichenic fungi origin, Terricollene A. The results were interpreted using root mean square deviation (RMSD) and radius of gyration (Rg).
The terricollene A-4AXX complex demonstrated the least deviations among all and followed a stable equilibrium throughout the process. The terricollene-3GQY complex displayed significant structural variations up to 10 ns; however, variations were observed to be lowered after that. Nonetheless, terricolleneA bound 2NZT complex experienced more significant fluctuation near 56,000 ps before attaining the equilibrium state. Moreover, the observed deviations were within 5 ˚A. The results showed that all complexes experienced protein backbone deviation and fluctuations within the simulation time frame and attained the equilibrium near 100000 ps (Fig. 1).

Fig 1. RMSD deviation of the complexes
Each receptor-ligand complex displayed a variation in Rg measurements. The deviation in Rg was observed to be least (within 2 ˚A) in terricollene A bound 4AXX complex suggesting its compactness to be linear. Slight variations were observed in terricollene A- 3GQY complex during the process of simulation. However, the terricollene A-2ZNT demonstrated increased deviations beyond 3.5 ˚A, which was seen to plummet after 50000 ps. Comprehensively, the structure complexes followed their path of trajectory without much variation, indicating their overall stability, and compactness (Fig. 2).

Fig 2. Rg fluctuation of the complexes
MM-PBSA of the binding free energies of terricollene bound 3GQY, 2ZNT, and 4AXX complexes were −34.946 kcal/mol, −13.2077 kcal/mol, and −18.011 kcal/Mol, respectively, and the overall energy of the complexes are depicted in Fig 3.

Fig 3. MM-PBSA calculation
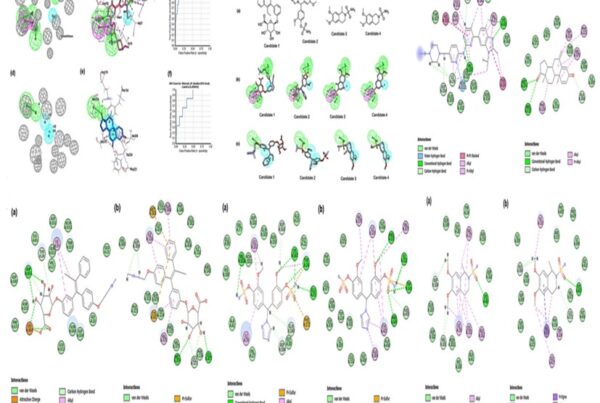
The final utilization of the interaction pharmacophore approach to investigate the structure-activity relationship (SAR) to distinguish active functional groups of chemical compounds shows the crucial pharmacophore at each drug target. This is illustrated in Fig 4. Hence, these integrated approaches bolstered them to identify the potential natural kinase inhibitors in a shorter period and cost-effective without any experimental studies.
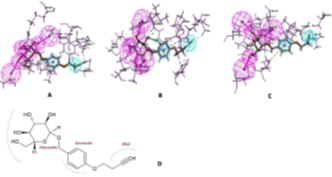
Fig 4. Pharmacophore features of terricollene A(D) concerning its binding interaction with the drug target (A, B & C)
SOLUTION: Intergarted screening approaches in BIOVIA suite
The impact of MD simulations in drug discovery and design has expanded significantly in the past few years, which can capture the structural behavior of protein and other biomolecular systems in full atomic detail. Establishing a stable and convergent simulation setup requires the correct assignment of protonation states to the receptors (amino acids), which is thought to be crucial for their catalytic activity and structural stability.
In addition, MD simulation has become very much substantial in optimizing the ligands for pharmaceutical applications. Also, the precision of results to check the stability of receptor-ligand interactions in the real-time environment with a SAR aided the researchers to solve the challenging scenario with limitations using BIOVIA Discovery Studio
Cite: S. Padhi, M. Masi, Y.K. Mohanta, M. Saravanan, S. Sharma, A. Cimmino, D. Shanmugarajan, A. Evidente, K. Tayung, A.K. Rai, In silico pharmacokinetics, molecular docking and dynamic simulation studies of endolichenic fungi secondary metabolites: An implication in identifying novel kinase inhibitors as potential anticancer agents, Journal of Molecular Structure, Volume 1273, 2023, 134390.
Acknowledgment:
We, the team thankful to Dr. S. Padhi and Dr. Amit Kumar Rai of IBSD (Institute of Bioresources and Sustainable Development) for sharing one of the research publications a case study.
Publication Date : December, 2022