Biovia Generative Therapeutics Design
Combine Human Ingenuity with AI For Faster Drug Discovery
BIOVIA Generative Therapeutics Design is a cloud-based integrated solution that optimizes drug design and discovery — potentially saving millions of research dollars:
- Combine advanced data science, machine learning, cheminformatics and structure-based modelling to explore chemical space employing Computational Chemistry Applications and Virtual Screening in Drug Discovery.
- Automate the virtual creation, testing and selection of novel small molecules utilizing Ligand-Based Drug Design and In Silico Analysis.
- Reduce the cost of physical testing by employing Molecular Modelling in Drug Design and Protein-Ligand Docking.
Enough Talk Let’s Build Something Together
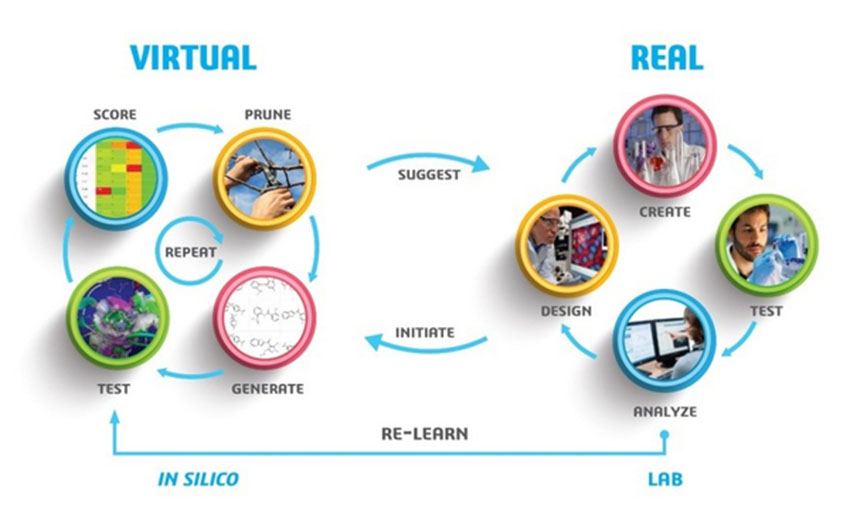
- Virtual cycles: Explore chemical space by “learning” from real experiments. The system virtually screens and optimizes candidate compounds using a combination of Machine Learning models and structure-based modelling and simulation methods, including Homology Modelling Stepsand Molecular Docking. Multi-objective optimization algorithms balance competing objectives, allowing teams to generate compounds that improve towards a TPP.
- Real cycles: Synthesize and test the most promising virtual compounds in the lab. Use the new data to improve predictive models and refine the exploration of chemical space. These V+R active learning cycles continue until you identify compounds that meet the TPP, employing 3D QSAR and ADMET Properties predictions.
- Modeling and Simulationmethods are essential for improving the quality of compound suggestions. Promising approaches include pharmacophore scoring, molecular docking and free energy methods for predicting binding affinities. BIOVIA has over 20 years of experience in supporting computational scientists using these methods, which we are integrating into our generative design solutions.
- Synthetic Feasibility scoring helps scientists to design virtually generated compounds with fewer experimental cycles, while significantly reducing overall discovery costs, incorporating Computational Chemistry Applications.
- V+R Analysis and Decision-Making pairs the unique expertise and intuition of the chemist with AI predictive modelling to shape the decision about which compound to make next Structure-Based Drug Designand Discovery Studio Visualizer. All virtual and real information is then combined in an analytics environment that takes into account preferred chemistries and reactions and available resources/knowledge from ‘prior art’ to find the most promising compounds to pursue.
- Cloud and Collaborative Platform– Molecular discovery is a team effort. Medicinal chemists work with biologists, computational scientists and data scientists. Many of these collaborations are now external. Common data models facilitate team collaboration and tech transfer to downstream activities. Hybrid Cloud, an integration between on-premises and cloud activities, ensures that the right people have access to the right information at the right time.